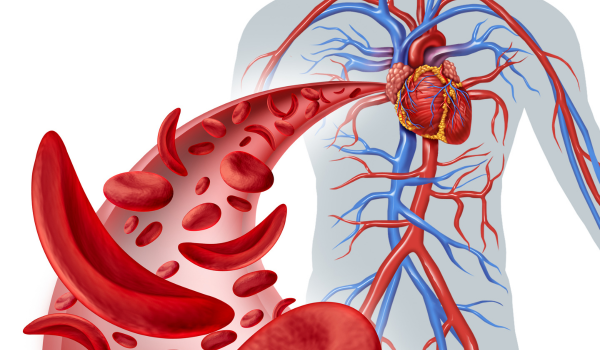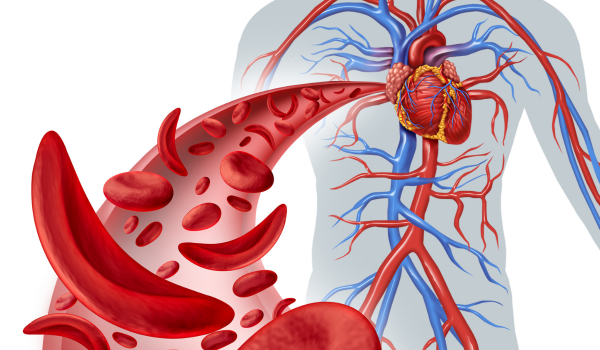
Sickle cell disease (SCD) is a group of inherited blood disorders that affect how red blood cells function. Instead of their usual round, flexible shape, red blood cells in people with SCD become stiff and crescent- or sickle-shaped. These cells have difficulty passing through blood vessels, which disrupts oxygen delivery to tissues and causes pain, organ damage, and other serious health issues. SCD is a lifelong condition, but advances in treatment and early diagnosis can significantly improve quality of life.
Types of SCD
Sickle cell disease comes in several forms, depending on the specific genes inherited from your parents:
-
HbSS (sickle cell anemia): The most severe and common form, caused by inheriting two sickle cell genes (one from each parent).
-
HbSC: Caused by inheriting one sickle cell gene and one hemoglobin C gene. It is typically less severe than HbSS.
-
HbS beta-thalassemia: A mix of sickle cell and beta-thalassemia traits. Beta-zero is more severe than beta-plus.
-
Rare types (HbSD, HbSE, HbSO): These result from combining the sickle cell gene with other abnormal hemoglobin variants.
Genetic testing can determine the specific type and help guide treatment decisions.
Common Symptoms
Symptoms of sickle cell disease can start as early as five to six months of age. Their severity and frequency vary widely from person to person:
-
Episodes of severe pain in the chest, joints, back, or abdomen
-
Fatigue and general weakness
-
Anemia due to rapid breakdown of red blood cells
-
Jaundice (yellowing of the eyes and skin)
-
Swollen hands and feet (especially in children)
-
Frequent infections due to spleen damage
-
Delayed growth and puberty
-
Vision problems or blindness
-
Priapism (painful, prolonged erections in males)
Pain crises, the hallmark symptom of SCD, can occur suddenly and last for hours or days, often requiring hospitalization.
Causes and Risk Factors
Sickle cell disease is inherited in an autosomal recessive pattern. A child must inherit a sickle cell gene from both parents to develop the disease:
-
Two copies (from both parents): Results in SCD.
-
One copy: The person is a carrier (sickle cell trait) but usually does not show symptoms.
Those of African, Hispanic, Middle Eastern, and South Asian descent have a higher prevalence of the sickle cell gene. If both parents carry the trait, there’s a 25% chance with each pregnancy that the child will have SCD.
Diagnosis Methods
SCD is typically diagnosed in infancy through newborn screening programs:
-
Heel-prick test: A blood sample is collected from the baby’s heel to detect abnormal hemoglobin.
-
Follow-up testing: Confirms the presence and type of SCD.
For older individuals or those with a family history of the condition, diagnosis may involve:
-
Blood tests: To examine hemoglobin types and quantities
-
Hemoglobin electrophoresis: To determine specific hemoglobin variants
-
Genetic testing: Especially helpful for parents planning to have children
Early diagnosis allows for prompt treatment and monitoring, which can prevent complications.
Available Treatments
Although there is no universal cure for SCD, several treatments can help manage symptoms and reduce complications:
Medications:
-
Hydroxyurea: Helps produce fetal hemoglobin and reduces pain episodes.
-
L-glutamine (Endari): Reduces the frequency of sickle cell crises.
-
Voxelotor (Oxbryta): Improves hemoglobin levels and reduces anemia.
-
Crizanlizumab (Adakveo): Prevents vaso-occlusive crises.
Pain Management:
-
Over-the-counter painkillers (acetaminophen, ibuprofen)
-
Prescription opioids for severe pain
-
Heating pads, hydration, and rest during pain episodes
Antibiotics and Vaccinations:
-
Daily penicillin in children to prevent infections
-
Routine immunizations to reduce risk of pneumonia and other illnesses
Blood Transfusions:
-
Increase healthy red blood cells
-
Reduce stroke risk in children
-
Used in emergencies or regularly depending on condition severity
Bone Marrow Transplant:
-
The only known cure for some individuals
-
Involves replacing damaged marrow with healthy donor cells
-
Best success with matched sibling donors
Gene Therapy:
-
Recently approved options modify genetic material to correct the underlying issue
-
Access is limited and may not be widely available for several years
Preventive Measures
Because SCD is inherited, it cannot be prevented after birth. However, steps can be taken to reduce symptom severity and avoid complications:
-
Genetic counseling: Before pregnancy to assess risk of passing on the gene
-
Prenatal testing: To detect the condition in the fetus early
-
Hydration and nutrition: Maintain healthy diet and fluid intake
-
Avoid extreme temperatures: Cold and heat can trigger crises
-
Exercise with caution: Avoid overexertion
-
Routine care: Regular checkups with hematologists and specialists
Early intervention can significantly improve health outcomes and prevent life-threatening complications.
Potential Complications
Sickle cell disease affects multiple organs over time, leading to various complications:
-
Acute chest syndrome: Blockage in lung blood vessels causing chest pain, coughing, and difficulty breathing
-
Stroke: Reduced blood flow to the brain
-
Kidney failure: Due to limited blood flow and cell damage
-
Liver problems: Overwork from removing damaged cells
-
Priapism: Painful, prolonged erections due to trapped blood
-
Avascular necrosis: Bone tissue death, especially in hips
-
Splenic sequestration: Spleen enlargement and sudden blood loss
-
Pulmonary hypertension: Elevated blood pressure in the lungs
-
Vision loss: Retinal damage due to poor circulation
Immediate medical attention is essential for any suspected complication.
Living with SCD
Managing sickle cell disease requires a holistic and proactive approach:
-
Education: Understanding your condition empowers better decision-making
-
Pain management plans: Work with your healthcare provider to develop strategies
-
Mental health support: Chronic illness can lead to stress and depression
-
School and work accommodations: Allow for flexibility and rest as needed
-
Peer and community support: Support groups offer emotional encouragement and shared experiences
-
Monitoring for long-term effects: Regular screenings for organ function, especially liver, kidney, lung, and eyes
In addition, travel and lifestyle planning are important. Individuals with SCD should be cautious when flying or visiting high-altitude locations, as low oxygen environments can trigger crises. Carrying medical ID and keeping essential medications on hand is strongly recommended.
Research and Future Outlook
The future of sickle cell disease care is promising thanks to innovations in medical research. Scientists are exploring:
-
Advanced gene-editing techniques: Such as CRISPR to correct defective genes
-
Improved transplant compatibility: Broadening access to bone marrow transplants
-
Personalized treatment plans: Using biomarkers to tailor therapies
-
Telemedicine and digital monitoring: Helping patients manage SCD from home
While many of these innovations are still in clinical trial stages, they represent hope for long-term, curative strategies.
Continued investment in awareness campaigns, community support, and equitable access to care will be key to improving outcomes for individuals living with sickle cell disease.